
OTHER LINKS

UCB Protein Structure Prediction Project

Sponsors:
 |
 |
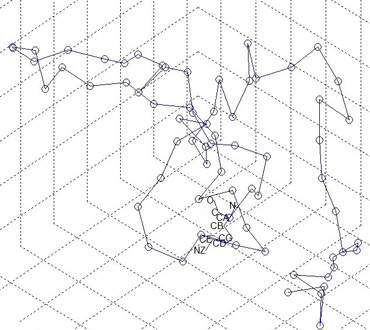
Figure 1: Protein 3D structure plotted in MATLAB with atoms on one residue labeled
Given the protein backbone structure, we aim to find side-chain conformations that minimize the protein structure's overall energy. Figure 1 shows a protein 3D structure with one side-chain (residue) labeled. Discrete side-chain conformations are known as rotamers. We predict side-chain conformation from a statistic perspective by using previously solved protein structures. Backbone-dependent rotamer libraries present rotamer conformations on the local backbone conformation as defined by the backbone dihedral angles Φ and Ψ. We have studied the 2010 backbone-dependent rotamer library by Dunbrack (http://dunbrack.fccc.edu/bbdep2010/) that uses an adaptive kernel on the backbone-dependent rotamer angle probability distributions. We have generated the smoothed rotamer probabilities based on the existing library, essentially replicating the work in Dunbrack’s paper. Figure 2 shows a smoothed rotamer probability of Χ 1 = g + for the residue type Serine. Using Dunbrack’s scoring function, which models energy components in the protein, we are developing a new sampling strategy, with the goal being to provide the same results with much less computation.
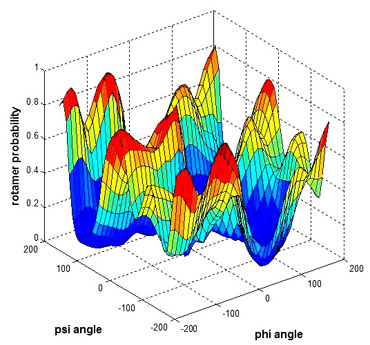
Figure 2: Smoothed rotamer probability of for the residue type Serine.